
Transposon mining, engineering and application
Transposon-mediated gene delivery and editing technology. The research directions include: (1) Transposon mining and engineering for efficient and safe gene delivery; (2) Transposon-encoded and RNA-guided tiny nucleases mining and engineering for efficient and safe gene editing; (3) Efficient and safe integration technology for large DNA fragment; (4) RIP mining and RIP chip developing in livestock. The research primarily utilizes model organisms such as E. coli, mammalian cells, yeast cells, zebrafish, mice, and pigs.
1. Transposon evolution and Biological importance
The transpositional properties of transposable elements enable their horizontal propagation, challenging the traditional understanding of vertical transmission of genetic material in the evolution of life. This characteristic allows genetic material to undergo horizontal transfer between different species, kingdoms, and domains. Deciphering the evolutionary patterns of TEs—including their origins, classification, horizontal transfer, domestication (as protein-coding genes), and adaptation (as gene-splicing elements and regulatory elements for expression control, such as enhancers, promoters, and lncRNAs)—is crucial. It sheds light on the role of transposon-mediated structural variations in reshaping functional genes, genomes, transcriptomes, and epigenomes, and deepens our understanding of how TEs contribute to phenotypic variation, genomic evolution, and species divergence.
2. Active transposon mining and engineering for efficient and safe gene delivery
The transposition properties of transposons enable them to mediate the transfer of large DNA fragments. Mining active transposons and developing efficient gene delivery tools through engineering optimization hold broad application prospects in human gene therapy and transgenic organism breeding.
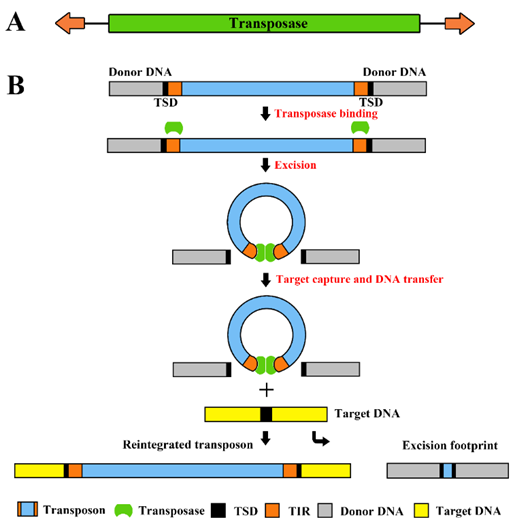
Fig.1. Structure of cut-and-paste transposons and their transposition mechanism. (A) The transposon is a mobile genetic element containing a transposase coding sequence (green box) flanked by terminal inverted repeats (TIRs; orange arrows on the left and right). (B) The transposase (green spheres) binds to its sites within the transposon TIRs (orange boxes). Excision takes place in a synaptic complex, and separates the transposon from the donor DNA (gray box). The excised element integrates into a target site in the target DNA (yellow box). This process generates target site duplications (TSDs, black boxes) which flank the newly integrated transposon. Most cut-and-paste transposons generate a transposon excision footprint in the donor DNA.
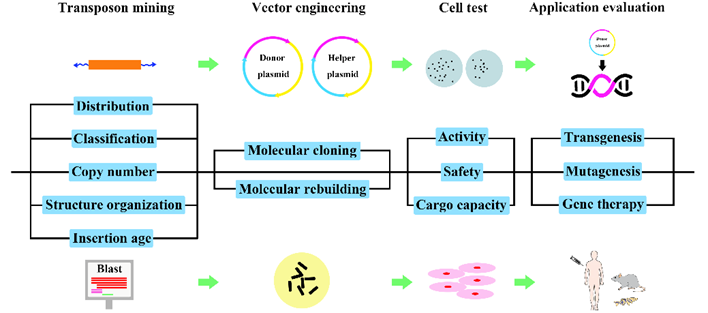
Fig.2. Experimental pipeline for the development of genetic tools based on active DNA transposons.
3. Transposon-encoded and RNA-guided tiny nucleases mining and engineering for efficient and safe gene editing
The Cas9 and Cas12a (Cpf1) nucleases, integral to the CRISPR/Cas gene editing system, possess substantial molecular weights, spanning 1200-1400 amino acids, which complicate their engineering, gene packaging, and delivery into cells. Additionally, the efficiency of gene editing is often suboptimal, and the prevalence of off-target effects considerably constrains the utility of CRISPR/Cas-based technologies in gene therapy and bio-breeding.
TnpB is a miniature nuclease associated with transposons. Research indicates that three types of transposons (IS605, IS607, IS1341) contain TnpB. It is an RNA-guided nuclease widely present in bacteria and archaea. In recent years, TnpB has garnered attention due to its similarity to the CRISPR-Cas system and is considered one of the evolutionary precursors of the CRISPR-Cas system. TnpB can process its own mRNA to produce guide RNAs. These guide RNAs are short RNA molecules that can direct the TnpB nuclease to specific DNA sequences. The processing involves cleaving the mRNA to generate guide RNAs, which are then used to localize the nuclease activity to the correct position in the genome.
Compared with large nucleases (Cas9 and Cas12a), compact RNA-guided nucleases offer substantial advantages, including easier synthesis, improved stability, and more efficient genetic manipulation and cellular transfection. Research and development of miniaturized RNA-guided endonucleases, particularly those originating from transposons and enhanced through artificial intelligence (AI) technologies, are crucial for increasing the safety and effectiveness of gene editing tools for human gene therapy and the development of genetically modified organisms.
4. Efficient and safe integration technology for large DNA fragment
Retroviruses and DNA transposons are frequently employed as gene therapy delivery systems. Retroviruses harness the inherent functionality of endogenous reverse transcriptase and transposition via a copy-and-paste mechanism, but this process can result in unstable retroviral insertions due to the involvement of RNA intermediates and reverse transcription. Furthermore, retroviral vectors present hurdles in manipulation and suffer from restricted payload capacity and high cost.
Conversely, DNA transposons operate through a direct cut-and-paste mechanism, facilitating a more predictable and robust genomic integration. Researchers have successfully developed potent gene delivery vectors, such as ZB, PS, Sleeping Beauty, and piggyBac, from DNA transposons for application in gene therapy, transgenesis, and mutagenesis. These vectors confer several benefits in gene therapy: low cost, substantial gene delivery capacity, stable integration, a diminished immune response, and manipulation simplicity with reduced risk of immunosuppression.
Nevertheless, gene therapy that involves DNA transposons, as well as retroviruses, carries the potential hazard of random transgene integration, heightening the risk of insertional mutagenesis. Such unspecific incorporation can inadvertently affect gene function, including the regulation of oncogenes and tumor suppressor genes, posing a profound safety challenge in gene therapy.
Therefore, advancing gene delivery systems by discovering and engineering targeting DNA transposons, with aid of artificial intelligence (AI) and other technologies, is imperative. These enhancements can mitigate the dangers of arbitrary integration, thereby substantially elevating the safety profile of DNA transposon-mediated gene therapy.
Additionally, the construction of site-specific integration technology for large DNA fragments through the fusion of transposase and compact RNA-guided nucleases is also a key research direction in the laboratory.
5. RIP mining and RIP chip developing in livestock
Since transposons can transfer between and within species, they can generate rich genomic structural variations like other mutation sources. Structural variations generated by transposons are also known as transposon insertion polymorphisms (TIP). Transposons are major components of the genomes of many model animals, accounting for around 55% of the zebrafish genome, 35% of the frog genome, 45% of the silkworm genome, and 40% of the pig genome. In addition, since transposon insertion fragments are usually large (generally more than 200 bp), and mostly contain functional elements (such as promoters and enhancers), TIPs generally have stronger genetic effects than SNPs. Transposon insertion can cause changes in gene function and activity by importing regulatory elements, changing gene splicing modes, or changing epigenetic regulation, thereby causing phenotypic variations. Furthermore, since TIPs are mediated by endogenous transposases, while SNPs are natural mutations, the mutation rate of TIPs (2.5×10−2) is higher than that of SNPs (1.0-1.8×10–8).
Therefore, transposons not only promote the differentiation of genomes among species, but also generate rich genetic diversity within species, playing an important role in the formation of species, varieties, subspecies, strains, and new traits. As a result, polymorphisms formed by transposon insertions are important new molecular markers, whose applications in genetic evolution research provide a new perspective for us to understand the evolution of higher organism genomes. At the same time, transposon insertion polymorphism molecular marker technology has become an important tool for research on biodiversity, genetic evolution and molecular breeding. Compared with traditional SNP markers, transposon insertion polymorphism (TIP) markers have the advantages of high mutation rate, good stability, large quantity, wide distribution in genome, high genetic effects, simple and fast detection methods, and low development costs. Therefore, the molecular marker mining based on transposon insertion polymorphism (TIP) on the genome, as well as the development of TIP chips, have higher application value in genetic breeding research such as QTL fine mapping and whole genome selection. Over 50% of the structural variations in the genomes of domestic animals, such as pigs, are generated by retrotransposon insertions mediated by retrotransposons, also known as Retrotransposon Insertion Polymorphism (RIP). Especially, the RIP markers produced by SINE retrotransposons, which are the most widespread and polymorphic transposons in livestock genomes, have the greatest potential for development.