From RepBase
TE introduction
Transposable element
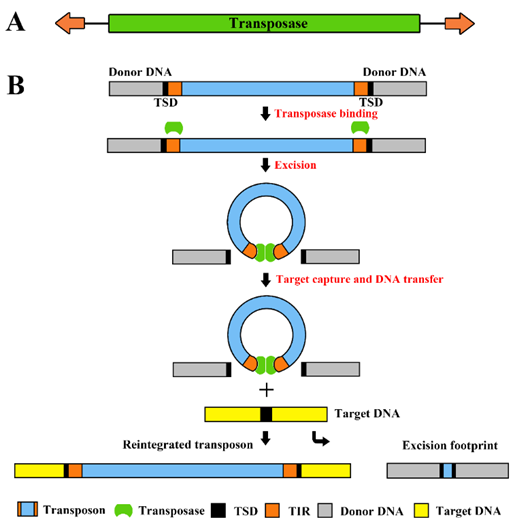
Transposable element (TE), also known as transposon or mobile element, is a diversity of DNA segments that can, in a process called transposition, move, or duplicate, from one location in the genome to another. The length of TEs ranges from less than 100-bp to more than 20-kb. After transposition, many types of TEs are flanked by short (~1–20 bp) direct repeats, target site duplications (TSDs), which are derived from the target sequence. However, some TE types, such as Helitron, several Harbinger families and CR1 retrotransposons, do not produce TSDs. The length of TSD is usually a characteristic of a group of TEs and its relatives, but may vary across different groups and superfamilies. TEs constitute the major part of repetitive sequences in most eukaryotic genomes. Other repetitive sequences include tandem repeats (satellite sequences or mini-satellite), sporadic genomic duplications and some multiple-copy host genes (such as rRNA, tRNA, histone genes, etc.). In fact, TEs can be viewed as intra-genomic parasitic elements. Similarly, inter-cellular virus can also be viewed as TEs since they can integrate into the host genome, such as the LTR-retrovirus. TEs have diverse evolutionary impact on host genome.
Machinery and classification of TEs
The transposition of TEs is largely depending on the diverse TE-encoded enzymatic machinery. Some identified enzymes or domains so far include reverse transcriptase, endonuclease, DD[E/D]-transposase (Tpase), Tyrosine-recombinase and Rep/Helicase1. Depending on whether reverse transcription is involved in transposition, TEs are classified into retrotransposon (Class I) and DNA transposon (Class II). In DNA transposons, the underlying enzymes could be either DD[E/D]-transposase (Tpase), or Rep/Helicase (for Helitron), or Tyrosine-recombinase (for Crypton). Depending on the sequence similarity of the key enzymes and additional DNA features, TEs are classified into superfamilies (see TE classification). Each superfamily includes numerous different families.
TE family
In the evolutionary history, a given TE, and its active copies, often maintain their activity in a relatively short period of time. Copies generated from them thus form a family. In older families, the TE members have undergone excessive degeneration, and barely represents the original active sequences. Therefore, family consensus sequences are often rebuilt from individual copies to represent the family. In genome sequences, TEs in most loci are only partial fragments, losing the transposition ability due to the lost of the functional element recognized by the transposition machinery. For example, most Non-LTR retrotransposons are 5’-truncated, due to the earlier termination of reverse transcription, and solo-LTR are likely the recombinant from two identical LTRs of LTR retrotransposons. Only a small percentage of TEs are complete in the sense of containing both termini and encode a full set of enzymes for its transposition. These TEs are termed as autonomous. Consequently, non-autonomous TE does not encode any enzyme involved in transposition, and are transposed in trans. Notably, except for these essential enzymes, TEs may occasionally capture some host genes or gene fragments in the long evolutionary history.
Weidong Bao, Ph. D.
TE classification
Historically, eukaryotic transposable elements are classified into two classes: Class I and Class II. Class I is Retrotransposon, which transposes through RNA intermediate. Class II is DNA transposon, which does not use RNA as a transposition intermediate. In other words, Class I includes all transposons encoding reverse transcriptase and their non-autonomous derivatives, while Class II includes all other autonomous transposons lacking reverse transcriptase and their non-autonomous derivatives. Another important information is that the genomes of prokaryotes (bacteria and archaea) does not contain any Class I transposons.
Class I transposon (retrotransposon)
Class I is subdivided into two large categories distinguished by the presence of long terminal repeats (LTRs): LTR retrotransposons and non-LTR retrotransposons. Recent studies revealed some other groups of eukaryotic retrotransposons, distinct from the above two by the transposition mechanism and/or the phylogeny of reverse transcriptase. They are DIRS retrotransposons (or tyrosine recombinase-encoding retrotransposons, YR retrotransposons) and Penelope-like retrotransposons (Penelope-like elements, PLE). To avoid the over-subclassification, in the classification implemented in Repbase, however, DIRS retrotransposons are included in LTR retrotransposons and Penelope-like retrotransposons are in non-LTR retrotransposons.
LTR retrotransposon contains LTRs at both ends, between which there are protein-coding regions. Proteins contain several catalytic domains: protease, reverse transcriptase, RNase H and integrase as well as structural proteins, called Gag and Env. LTR retrotransposons mobilize through reverse transcription of their own mRNA as a template, catalyzed by reverse transcriptase. cDNA is generated as an extrachromosomal DNA and it is then integrated into the genome by integrase. Integrase of LTR retrotransposons shows similarity to the transposase of some DNA transposons, especially Ginger1 and Ginger2 superfamilies, indicating the composite origin of LTR retrotransposons. LTR retrotransposons are subdivided into 5 superfamilies: Copia, Gypsy, BEL, DIRS and endogenous retroviruses (ERV). ERVs are retroviruses that omit their extracellular life style and replicate themselves in germ cells. ERVs are further divided into 5 groups, ERV1, ERV2, ERV3, ERV4 and endogenous lentivirus (ELV), but based on the classification of infectious (exogenous) retroviruses, which are classified into 8 genera, ERVs can be classified into more groups. ERV1 corresponds to two retroviral genera, Gammaretrovirus and Epsilonretrovirus, and ERV2 corresponds to Alpharetrovirus and Betaretrovirus. ERV3 and ERV4 do not have a corresponding infectious retrovirus group. International Committee on Taxonomy of Viruses (ICTV) classified some LTR retrotransposons as viruses: family Pseudoviridae for Copia, and family Metaviridae for Gypsy and BEL,
Based on the phylogeny of reverse transcriptase, besides Retrovirus, LTR retrotransposons are related to two viral families, Hepadnavirus and Caulimovirus, and both of them are occasionally integrated into the genomes. Plant Caulimoviruses are often integrated into the genome and Repbase has a category for them (Integrated Virus – Caulimoviridae).
Non-LTR retrotransposons lack LTRs, and usually they have poly A or simple repeats at their 3’-terminus. Non-LTR retrotransposons encode either of three types of endonucleases, restriction-endonuclease-like (RLE), apurinic-endonuclease-like (APE), or GIY-YIG endonuclease. Dualen is an exception that encodes both RLE and APE. Endonuclease nicks one strand of DNA and reverse transcriptase initiates reverse transcription using the exposed 3’ end as a primer and mRNA of non-LTR retrotransposon as a template. This mechanism is called Target-Primed Reverse Transcription (TPRT). TPRT is also used as a mechanism of integration of group II self-splicing introns, which also have reverse transcriptase. Group II intron is absent in eukaryotic nuclear genomes, and thus Repbase does not have group II intron entries.
Non-LTR retrotransposons are classified into groups (CRE, R2, Dualen/RandI, Ambal, L1, RTE, I and CR1) and further divided into many clades. The classification “clade” was first proposed by Malik and Eickbush, 1999, and now more than 30 clades have been proposed, which makes the classification of non-LTR retrotransposons complicated. GIRI serves a simple classification tool RTclass1, which is based on the neighbor-joining tree and the reference non-LTR retrotransposons. As of December 2016, Repbase contains 32 clades (CRE, NeSL, R4, R2, Hero, RandI/Dualen, L1, Proto1, Tx1, Proto2, RTE, RTEX, RTETP, I, Nimb, Ingi, Vingi, Tad1, Loa, R1, Outcast, Jockey, CR1, L2, L2A, L2B, Kiri, Rex1, Crack, Daphne, Ambal, Penelope) in its classification.
Non-autonomous non-LTR retrotransposons show composite structures and they are called short interspersed elements or SINEs, corresponding to long interspersed elements or LINEs, the synonym of autonomous non-LTR retrotransposon. SINEs are classified into 5 groups in Repbase based on the origin of 5’ part of SINEs. SINE1 for 7SL RNA, SINE2 for tRNA, SINE3 for 5S rRNA, SINEU for U1 or U2 snRNA and SINE4 for unknown origin. Another way of classification of SINEs is based on the similarity of their central regions. CORE-SINE, V-SINE, Deu-SINE (or Nin-SINE), Ceph-SINE and Meta-SINE are proposed, although Repbase does not use this classification since it contradicts the classification based on the origin of 5’ regions. Some entries have these classification terms in their keyword section.
Class II transposons (DNA transposons)
Repbase contains 23 superfamilies of Class II as of December 2016. Among them, Helitron, Polinton and Crypton show distinct features from other DNA transposons. 18 superfamilies (Mariner/Tc1, hAT, MuDR, EnSpm/CACTA, piggyBac, P, Merlin, Harbinger, Transib, Polinton, Kolobok, ISL2EU, Sola, Zator, Ginger1, Ginger2/TDD, IS3EU and Dada) encode a D-D-D/E-type integrase/transposase for their catalytic reaction of integration. This integrase shares the features of catalytic core with integrase of LTR retrotransposons, and especially, Ginger1, Ginger2/TDD and Polinton superfamilies likely share the origin with LTR retrotransposons. The catalytic cores of Academ, Novosib and Zisupton are less characterized and there still remains a possibility that they encode a protein not related to D-D-D/E-type integrase. It is noteworthy that because the sequence is extremely divergent except the catalytic residues, the presence of conserved D-D-D/E core sequence does not guarantee their common origin; they may have independently evolved. Integrase is featured with RNase H-fold based on its ternary structure.
Notably, the scope and sub-structures of certain superfamilies are not fixed, and new groups could emerge. For example, previously, Harbinger and ISL2EU show similarity to known prokaryotic DNA transposons, IS5 family. Recently, 3 other sibling groups were recognized: Spy, Nuwa and Pangu. The latter three names are not appearing yet in Repbase, but could be combined with the other two groups under one chosen name, such as Harbinger, in the future.
EnSpm/CACTA and Transib share some residues between the second conserved D and E. Two superfamilies, Mirage and Chapaev, which were previously present in the classification in Repbase, are now integrated into EnSpm/CACTA based on their similarity.
MuDR, P, hAT, Kolobok, and Dada share the motif C/DxxH between the second conserved D and E. Rehavkus, previously present in Repbase as a superfamily, was integrated into MuDR. It is reported that MuDR shows similarity to the prokaryotic IS256 family.
Tc1/Mariner and Zator are related to prokaryotic IS630 family. Merlin is related to prokaryotic IS1016 family. Integrases of Ginger1, Ginger2/TDD, Polinton, as well as LTR retrotransposons are related to those of prokaryotic IS3/IS481 family. Sola is subdivided into three groups, Sola1, Sola2 and Sola3.
Crypton encodes a tyrosine recombinase, which is also characterized in some prokaryotic DNA transposons and DIRS retrotransposons. Crypton is subdivided into several groups (CryptonA, CryptonF, CryptonI, CryptonS and CryptonV), which may or may not share the common ancestor in eukaryotes; they may have independently evolved from prokaryotic DNA transposons.
Helitron encodes a protein including helicase and HUH nuclease, and recently the transposition mechanism of Helitron was characterized by experimental analysis. Helitron can be subdivided into two groups, Helitron1 and Helitron2, though Repbase have not implemented this classification yet.
Polinton, also called Maverick, is expected to transpose similarly with other DNA transposons, but it likely generates extrachromosomal DNA and replicate by themselves using the encoded DNA polymerase. Recently Polinton is proposed to be a genome-integrated endogenous virus, and its viral form is designated Polintovirus.
Repbase also contains other types of repeats, such as Satellite Repeats (SAT) and Microsatellites (MSAT), Multicopy genes (rRNA, tRNA, snRNA), Integrated Virus (DNA Virus and Caulimoviridae) and uncharacterized repeat sequences (Repeats). Please note that Repbase entries for multicopy genes are just representatives and do not always correspond to a specific functional copy or an obvious
Kenji K. Kojima, Ph. D.
TE identification and mapping
Given the huge diversity of TEs in most eukaryotic genomes and the lack of sequence conservation across species, it is often a challenge to mine TE sequences from a genome (mapping or detection). To some extent, this task can be done with blasting against a reference TE library (Repbase database) by Censor or RepeatMasker. For new genome sequences, it is necessary to perform TE mining or discovery from a scratch. A plenty of computing tools have been developed for this goal. Based on the underlying approaches, these tools can be divided into three categories. The first group are based on the homology or repetitiveness of TEs, such as RepeatScout and Recon. The second group are based on the structure features of certain types of TEs. The third group are based on comparative genomic method. Different tools have their own limits in performance. Some efforts attempt to combine multiple approaches, such as REPET and RepeatModeler. Up to now, a common problem of these automate tools is that a significant number of the output sequences are flawed in sequence accuracy and annotation. Manual curation is often required to get the full-length, high-quality family consensus. The workflow adopted in Repbase is illustrated below.
Weidong Bao, Ph. D.
Impact on genome
Diseases
Transposition of transposable elements into or near a gene may cause deleterious effects by disrupting the protein coding sequence or altering the gene expression or regulation. Many genetic diseases and cancers caused by transposition of transposable elements have been reported. The presence of transposable elements may have a fundamentally negative effect on individual genomes, however, they cannot be neglected when considering the genome evolution.
Genome size
Beside the whole genome duplications, transposable elements also account for the genome size variation. Some studies on closely related species have indicated that such genome size difference can be explained by some recent massive multiplication of transposable elements.
Domestication and Exaptation
TEs can serve as sources of host genes. A significant number of human genes are partly originated from transposable elements. The most famous example is the recombination activating gene 1 (rag1), whose protein catalyzes V-D-J recombination of immunoglobulin gene, which is the main contributor to the variety of antibody. The gene rag1 is originated from a DNA transposon belonging to Transib superfamily. Another group of DNA transposons, Cryptons, contribute to at least 6 human genes, among which 4 are paralogous. Retrotransposons, as well as DNA transposons, have contributed to human gene repertoire. Syncytin 1 is derived from an envelope protein gene of endogenous retrovirus, and contributes to the formation of placenta. The gene peg10 was originated from a Gypsy superfamily of LTR retrotransposons, and the methylation status around this gene is silenced only on paternally inherited chromosome and controls the fetus development. These events are called “molecular domestication”, where the whole or a part of transposons is integrated with the system of their host organism. The term “exaptation” specifically indicates the events where a part of transposons is incorporated with their host system, but not as a part of protein-coding gene. TEs can act as promoter, enhancer, insulator, intron splice site, polyadenylation signal, or any other function in the cell. One example is an enhancer derived from an ancient SINE element, LF-SINE, which controls the expression of ISL1 in the limbs of tetrapods. Many other examples are reported that TEs are the main contributor of ultraconserved elements (UCEs), and are highly conserved in sequence along very long time scale. It is revealed that ancient transposable elements are going to be concentrated in UCEs.
Carrier subpopulation (CASP) hypothesis
GIRI’s team headed by the late Jerzy Jurka proposed a hypothesis called “Carrier subpopulation (CASP)”. It can explain the different repertoires of active transposon families in each species. During speciation, a large population is split into many small subpopulations. In each subpopulation, different active families of transposons multiplicate and are fixed at random due to genetic drift. In a small subpopulation, slightly disadvantageous mutations, such as transposition of transposable elements, can be fixed. It suggests that a new copy of transposable elements can be fixed without any immediate benefit. After the fixation, copies of transposable elements can be eliminated or domesticated/exapted.
Kenji K. Kojima, Ph. D.